
医疗器械单一审核方案(Medical Device Single Audit Program ,缩写为MDSAP)是由国际医疗器械法规论坛IMDRF发起,以ISO 13485为基础,结合五个参与国(美国、加拿大、巴西、澳大利亚和日本)的法规要求,由认可的审核机构进行一次审核满足多国法规要求的方案。审核周期通常为3年。
MDSAP审核由具备授权资质的审核机构进行,例如BSI(英国标准协会British Standards Institution)。
如果制造商的医疗器械要出口到以下5个国家,就可以考虑MDSAP。
| 序号 | 国家 | 监管机构 | 备注 |
| 1 | 澳大利亚 | 澳大利亚药品管理局(TGA) | 澳大利亚药品管理局(TGA)使用MDSAP的审核报告作为评估市场准入要求的一部分。可豁免上市批准要求的医疗器械,或有特殊政策要求的情况除外。 |
| 2 | 巴西 | 巴西国家卫生监督局(ANVISA) | 巴西国家卫生监督局(ANVISA)采用MDSAP审核报告和结果作为产品上市前和上市后审核的重要输入,并作为法规技术评审的支持要素。对III类和IV类的医疗器械,制造商可以用MDSAP审核去替代ANVISA的审核去获取NAVISA的GMP证书,从而获得市场准入。 |
| 3 | 加拿大 | 加拿大卫生部(HC) | 加拿大卫生部(HC)宣布,从2019年1月1日起,MDSAP将强制替代CMDCAS成为加拿大医疗器械准入审核方案。未在2019年1月1日前获得MDSAP证书将意味着您的医疗器械产品不能在加拿大销售。 |
| 4 | 日本 | 日本厚生劳动省(MHLW) >日本医药医疗器械管理局(PMDA) | 日本厚生劳动省(MHLW)以及医药医疗器械管理局(PMDA)对于产品上市前和上市后的审核,都可使用MDSAP报告。 |
| 5 | 美国 | 美国食品和药品管理局器械和放射健康中心(FDA CDRH) | 美国食品和药品管理局器械和放射健康中心(FDA CDRH)宣布,MDSAP可替代FDA的常规检查(通常两年一次)。有因检查(For Cause)、符合性后续审核(Compliance Follow-up)和上市前审批(PMA)产品除外 |
以下是审核的大致流程:
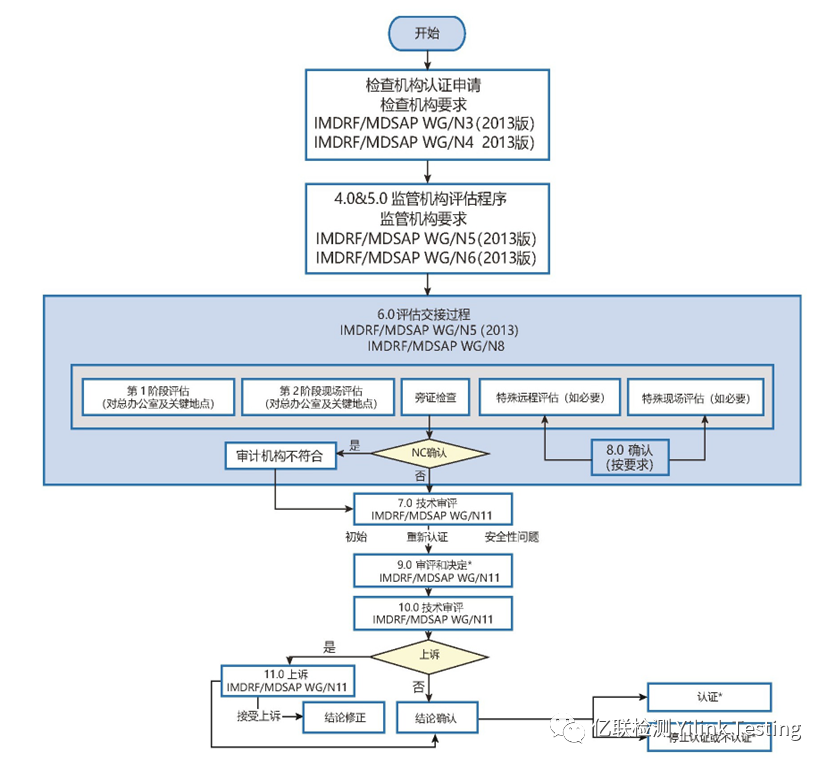
通过MDSAP审核不仅能为医疗器械制造商减轻多重法规审核的负担和干扰,减少处理多重审核发现所耗用的时间和资源,与多次独立审核相比,还大大降低了审核成本。
篇幅有限,如果需要了解更详细的内容,欢迎私信小亿交流。
发表回复